PanMiRa: Pan-cancer MiRNA-target Association
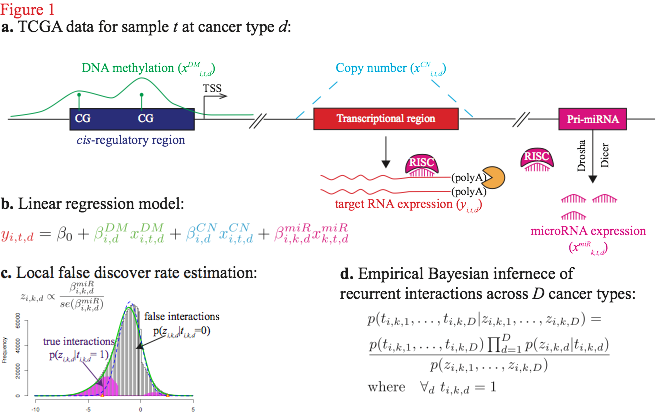
To reliably infer the distribution of the recurrent miRNA-target interactions implicated in the pan-cancer data panel, we developed a novel Bayesian framework called PanMiRa (Pan-cancer MiRNA-target Associations). Fig. 1 below illustrates the concept of the proposed model (details are in manuscript). Suppose the target expression level is a function of DNA methylation (DM), copy number (CN), and miRNA regulation. To estimate the individual miRNA-target relationships, we employed a multivariate linear regression model, which was fitted to the observed target expression, taking into account the biases due to the baseline expression level, DM, and CN effects (Fig. 1b). The resulting linear coefficient is indicative of the corresponding miRNA-target repression in the specific cancer type: the more negative it is the more likely the interaction is real. To translate the numerical coefficients into the language of probability, we first transformed the coefficients into $z$-scores and then estimated the corresponding local false discovery rate ($locfdr$) using the procedure developed by Efron (2004) (Fig. 1c). The technique was invented for large-scale simultaneous hypothesis testings by estimating the null distribution directly from the test statistics. The resulting local FDR can be considered as an empirical Bayes version of the FDR derived from the popular Benjamini-Hochberg method and is equivalent to the posterior probability of false interactions given the z-scores.
The empirical distribution estimated from the locfdr approach is essentially the likelihood of z-scores given the underlying interaction status and enables estimating the joint posterior of the true interactions across all of the 12 cancer types via an empirical Bayes algorithm (Chen et al., 2013) (Fig. 1d). For each interaction, we first assume a uniform prior for all possible combinations of its binary value over the 12 cancer types. In particular, each interaction can take on 2^12=4096 possible combinations, and the initial prior is 1/4096 for each combination. Using Bayes rule and assuming the interactions at different cancer types are conditionally independent given their true interaction status, we can compute the posterior for each combination and re-estimate the prior by marginalization of the joint posteriors. We then alternate between the prior and posterior inference steps until little change occurs between the likelihoods at the current and previous iteration. The posteriors of interest associated with recurrent interactions correspond to the particular combination, where the interaction is positive across all of the 12 cancer types.
‣ PanMiRa source code:
-
•panmira_source.zip (3 KB)
-
•The above zip contains the scripts for the PanMiRa model and the script for calculating the z-scores as discussed in the paper.
‣ Pan-cancer data panel corresponding 12 distinct cancer types from TCGA
-
•tcga_join.RData (864.3 MB)
-
•The above data used in the paper is in RData format, which can be loaded directly in R.
‣ R source code, data, and results in one ZIP archive
-
•panmira.zip (3.2 GB)
References:
-
•Efron, B. (2004). Large-Scale Simultaneous Hypothesis Testing. Journal of the American Statistical Association, 99(465), 96–104. doi:10.1198/016214504000000089
-
•Chen, X., Slack, F. J., & Zhao, H. (2013). Joint analysis of expression profiles from multiple cancers improves the identification of microRNA-gene interactions. Bioinformatics (Oxford, England). doi:10.1093/bioinformatics/btt341