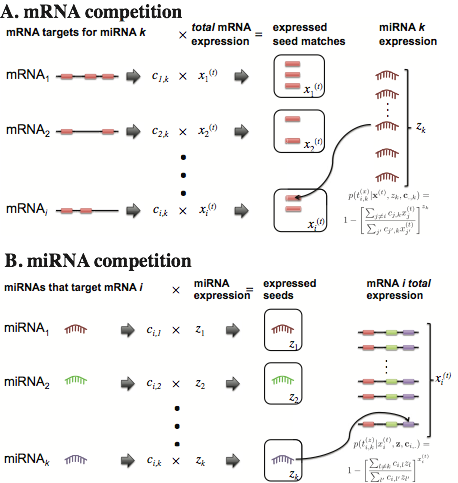
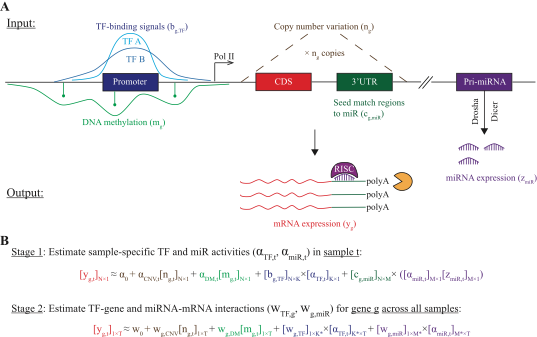
ProMISe operates in two phases by inferring the probability of mRNA (miRNA) being the targets ("targets") of miRNA (mRNA), taking into account the expression of all of the mRNAs (miRNAs) due to their potential competition for the same miRNA (mRNA). Due to mRNA transcription and miRNA repression events simultaneously happening in the cell, Roleswitch assumes that the total transcribed mRNA levels are higher than the observed mRNA levels at the equilibrium and iteratively updates the total transcription of each mRNA targets based on the above inference.
-
Citation:
-
Li, Y., Liang, C., Wong, KC, Jin, K., and Zhang, Z. (Feb, 2014) Inferring probabilistic miRNA-mRNA interaction signatures in cancers: a role-switch approach. Nucleic Acids Research, 42(9), e76. doi: 10.1093/nar/gku182
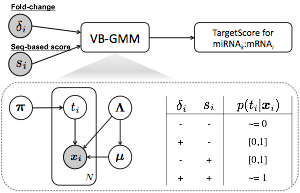
TargetScore is used to infer the posterior distributions of microRNA targets by probabilistically modeling the likelihood microRNA-overexpression fold-changes and sequence-based scores. Variational Bayesian Gaussian mixture model (VB-GMM) is applied to log fold-changes and sequence scores to obtain the posteriors of latent variable being the miRNA targets. The final targetScore is computed as the sigmoid-transformed fold-change weighted by the averaged posteriors of target components over all of the features.
-
Citation:
-
Li, Y., Goldenberg, A., Wong, KC., Zhang Z. (Oct, 2013). A probabilistic approach to explore human miRNA targetome by integrating miRNA-overexpression data and sequence information. Bioinformatics (Oxford, England), 30(5), 621–628. doi:10.1093/bioinformatics/btt599
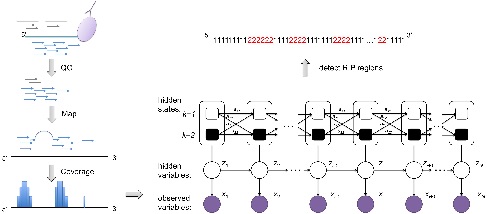
Infer and discriminate RIP peaks from RIP-seq alignments using two-state HMM with negative binomial emission probability. While RIPSeeker is specifically tailored for RIP-seq data analysis, it also provides a suite of bioinformatics tools integrated within this self-contained software package comprehensively addressing issues ranging from post-alignments processing to visualization and annotation. You can download and install RIPSeeker from Bioconductor.
-
-
Citation:
-
Li, Y., Zhao, D. Y., Greenblatt, J. F., & Zhang, Z. (March, 2013). RIPSeeker: a statistical package for identifying protein-associated transcripts from RIP-seq experiments. Nucleic Acids Research, 41(8), e94. doi:10.1093/nar/gkt142
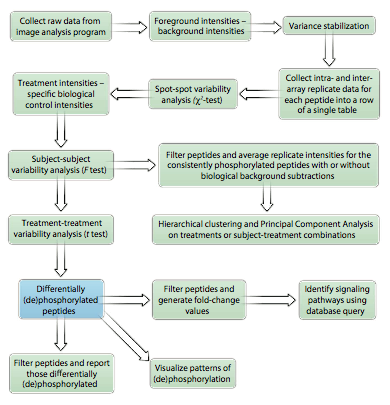
PIIKA: Platform for Integrated, Intelligent Kinome Analysis
PIIKA is a software pipeline implemented in R for analyzing data from kinome microarrays. I do not maintain this software. If you wish to use PIIKA for academic, non-commercial purposes, please e-mail here to obtain a license form.
-
Citation:
-
Li, Y., Arsenault, R. J., Trost, B., Slind, J., Griebel, P. J., Napper, S., and Kusalik, A. (April, 2012). A Systematic Approach for Analysis of Peptide Array Kinome Data. Science Signaling, 5(220), pl2–pl2.