
Home
About Us
Description
Updates
Help Conquer Cancer
Our goal is to improve the results of protein X-Ray crystallography. Improving the protein crystallography pipeline will enable researchers to determine the structure of many cancer-related proteins faster. This will lead to improving our understanding of the function of these proteins, and enable potential pharmaceutical interventions to treat this deadly disease.
If you have not joined yet (and you run Windows, Linux or Mac), you can become a member at World Community Grid. We would welcome if you join our Integrative Discovery Team after registering. However, any computing resources you donate to one of the five active projects at WCG is highly appreciated and will greatly help with these important scientific computations; and we too compute on multiple projects: .
More information about our project is available at Help Conquer Cancer.
If you have not joined yet (and you run Windows, Linux or Mac), you can become a member at World Community Grid. We would welcome if you join our Integrative Discovery Team after registering. However, any computing resources you donate to one of the five active projects at WCG is highly appreciated and will greatly help with these important scientific computations; and we too compute on multiple projects: .
More information about our project is available at Help Conquer Cancer.


Igor Jurisica
Principal Investigator
University Health Network
Principal Investigator
University Health Network

Christian Anders Cumbaa
Research Associate
University Health Network
Research Associate
University Health Network

George De Titta
Principal investigator
Co-Director of the HTS laboratory
Hauptman-Woodward Medical Research Institute
Principal investigator
Co-Director of the HTS laboratory
Hauptman-Woodward Medical Research Institute

Joseph R. Luft
Principal investigator
Co-Director of the HTS laboratory
Hauptman-Woodward Medical Research Institute
Principal investigator
Co-Director of the HTS laboratory
Hauptman-Woodward Medical Research Institute

Edward H. Snell
Principal investigator
Hauptman-Woodward Medical Research Institute
Principal investigator
Hauptman-Woodward Medical Research Institute

Michael Malkowski
Principal Investigator
Hauptman-Woodward Medical Research Institute
Principal Investigator
Hauptman-Woodward Medical Research Institute
Help Conquer Cancer
The mission of Help Conquer Cancer is to improve the results of protein X-Ray crystallography, which helps researchers not only annotate unknown parts of the human proteome, but importantly improves their understanding of cancer initiation, progression and treatment.
Significance
In order to significantly impact the understanding of cancer and its treatment, novel therapeutic approaches capable of targeting metastatic disease (or cancers spreading to other parts of the body) must not only be discovered, but also diagnostic markers (or indicators of the disease), that can detect early stage disease, must be identified.
Researchers have been able to make important discoveries when studying multiple human cancers, even when they have limited or no information at all about the involved proteins. However, to better understand and treat cancer, it is important for scientists to discover novel proteins involved in cancer, and their structure and function.
Scientists are especially interested in proteins that may have a functional relationship with cancer. These are proteins that are either over-expressed or repressed in cancers, or proteins that have been modified or mutated in ways that result in structural changes to them.
Improving X-Ray crystallography will enable researchers to determine the structure of many cancer-related proteins faster. This will lead to improving our understanding of the function of these proteins, and enable potential pharmaceutical interventions to treat this deadly disease.
Researchers have been able to make important discoveries when studying multiple human cancers, even when they have limited or no information at all about the involved proteins. However, to better understand and treat cancer, it is important for scientists to discover novel proteins involved in cancer, and their structure and function.
Scientists are especially interested in proteins that may have a functional relationship with cancer. These are proteins that are either over-expressed or repressed in cancers, or proteins that have been modified or mutated in ways that result in structural changes to them.
Improving X-Ray crystallography will enable researchers to determine the structure of many cancer-related proteins faster. This will lead to improving our understanding of the function of these proteins, and enable potential pharmaceutical interventions to treat this deadly disease.
X-Ray Crystallography
One of the favored methods for protein-structure determination is X-ray crystallography. Through this method, scientists use the high-throughput crystallization pipeline to help not only annotate unknown parts of the human proteome, which in turn will help to improve our understanding of cancer initiation, progression and treatment. (NOTE: There are other approaches to understanding the structure and function of proteins, including the Human Proteome Folding Project also running on World Community Grid. Given the essential nature of this work, it’s important to advance every research technique to complete our understanding of the human organism and disease.)
There are two main steps involved in X-ray crystallography:
Crystallizing the protein is not a straightforward procedure. There are many thousands of possible conditions that affect the process (concentration of a protein and solution, temperature, pH, chemical additives, etc.), but scientists must find the appropriate combination of these conditions for a protein to crystallize. For example, with sugar, if you change the water to another liquid, change the temperature or concentrations, you may not get a crystal. Similarly, for a given protein, the challenge is to know what conditions will lead to forming a crystal – what solution, what temperature, pH, etc.
The resultant protein crystal also must be well-formed and large enough in order for x-rays to detect the protein’s structure at high resolution. If the conditions are not perfect for crystallizing the protein, the process can result in either a micro-crystal, which is too small for the protein’s structure to be determined; a precipitate, which shows some changes, but does not lead to crystallization event directly; or no change may have occurred at all.
Frustrating the situation is that, as yet another barrier to progress, usually the more important the protein is to cancer research, the harder that protein is to crystallize. Many proteins involved in cancer are long chains, or they require additional proteins to properly fold and cannot be crystallized by themselves.
In order to run the millions of combinations necessary to successfully crystallize a protein, scientists have used robots to perform the work. Robots are able to put in place the various crystallization conditions faster and more accurately. To further facilitate the process, result of each of the millions of crystallization experiments are photographed.
Currently, scientists at Hauptman-Woodward Medical Research Institute in Buffalo (HWI) have run more than 86 million crystallography experiments for more than 9,400 proteins. As a result, they have 86 million pictures of these proteins that have gone through the X-ray crystallography high-throughput screening pipeline. Each of these pictures needs to be analyzed to determine what the result of the experiment is – i.e., crystal, precipitate, phase separation, skin effect, no change.
One of the challenges is the tremendous size of these datasets, which requires over 25 TB of storage (or equivalent to more than 9,000 DVDs). IBM’s Blue Gene supercomputer has provided assistance in this phase of the work, by running a special image compression algorithm to reduce the size of these images, without losing content. The other challenge is to comprehensively analyze an image to determine the crystallization outcome, a task that requires approximately 10 hours to process on a single computer. Researchers would thus require almost 100,000 years to analyze the existing pictures.
There are two main steps involved in X-ray crystallography:
- Crystallizing the protein. Although a lot more complex, this is similar to putting sugar into a cup of water and letting it sit for a while. Once the water evaporates, tiny sugar crystals appear.
- Sending X-rays through the crystal and depending on how they diffract, a mathematical model is used to determine and observe the protein’s structure.
Crystallizing the protein is not a straightforward procedure. There are many thousands of possible conditions that affect the process (concentration of a protein and solution, temperature, pH, chemical additives, etc.), but scientists must find the appropriate combination of these conditions for a protein to crystallize. For example, with sugar, if you change the water to another liquid, change the temperature or concentrations, you may not get a crystal. Similarly, for a given protein, the challenge is to know what conditions will lead to forming a crystal – what solution, what temperature, pH, etc.
The resultant protein crystal also must be well-formed and large enough in order for x-rays to detect the protein’s structure at high resolution. If the conditions are not perfect for crystallizing the protein, the process can result in either a micro-crystal, which is too small for the protein’s structure to be determined; a precipitate, which shows some changes, but does not lead to crystallization event directly; or no change may have occurred at all.
Frustrating the situation is that, as yet another barrier to progress, usually the more important the protein is to cancer research, the harder that protein is to crystallize. Many proteins involved in cancer are long chains, or they require additional proteins to properly fold and cannot be crystallized by themselves.
In order to run the millions of combinations necessary to successfully crystallize a protein, scientists have used robots to perform the work. Robots are able to put in place the various crystallization conditions faster and more accurately. To further facilitate the process, result of each of the millions of crystallization experiments are photographed.
Currently, scientists at Hauptman-Woodward Medical Research Institute in Buffalo (HWI) have run more than 86 million crystallography experiments for more than 9,400 proteins. As a result, they have 86 million pictures of these proteins that have gone through the X-ray crystallography high-throughput screening pipeline. Each of these pictures needs to be analyzed to determine what the result of the experiment is – i.e., crystal, precipitate, phase separation, skin effect, no change.
One of the challenges is the tremendous size of these datasets, which requires over 25 TB of storage (or equivalent to more than 9,000 DVDs). IBM’s Blue Gene supercomputer has provided assistance in this phase of the work, by running a special image compression algorithm to reduce the size of these images, without losing content. The other challenge is to comprehensively analyze an image to determine the crystallization outcome, a task that requires approximately 10 hours to process on a single computer. Researchers would thus require almost 100,000 years to analyze the existing pictures.
World Community Grid and "Help Conquer Cancer"
Using the power of World Community Grid, scientists at the Ontario Cancer Institute, Princess Margaret Hospital, and the University Health Network will process the existing 86 million images of proteins that have been screened in the high-throughput crystallization pipeline at the HWI in Buffalo. World Community Grid will run a CrystalVision program that the researchers at OCI have developed to analyze the features of individual images to determine the outcome of the crystallization screen – crystal, micro crystal, phase separation, skin, or a precipitate, or if no change occurred.
If a crystal occurs, crystallographers can put the protein through the optimization process to determine the optimal conditions for the crystallization, and in turn perform a diffraction experiment to determine the structure of the protein. What’s more, scientists can compare proteins that have successfully crystallized against proteins of unknown structure that have similar characteristics, based on the results from the crystallization screen. This can be the starting point for crystallization for these proteins so that their structure can be determined.
If the crystal produced was not well-formed or large enough, scientists can still use the information to help them better determine the conditions necessary to create a well-formed crystal. For example, they may learn that Protein X and Condition A resulted in a micro crystal, and Protein A and Condition Z resulted in a micro crystal as well. Based on this information, they can then run additional experiments to deduce what conditions need to be optimized to create a larger and well-formed crystal.
Analyzing the results from this experiment will also lead to better understanding the underlying principles of protein crystallography. For the first time, a comprehensive crystallography image analysis will be done, which was impossible before due to computational complexity. In turn, CrystalVision will be improved to provide faster and more accurate image classification.
Improving the protein crystallography pipeline will enable researchers to determine the structure of many cancer-related proteins faster. This will lead to improving our understanding of the function of these proteins, and enable potential pharmaceutical interventions to treat this deadly disease.
If a crystal occurs, crystallographers can put the protein through the optimization process to determine the optimal conditions for the crystallization, and in turn perform a diffraction experiment to determine the structure of the protein. What’s more, scientists can compare proteins that have successfully crystallized against proteins of unknown structure that have similar characteristics, based on the results from the crystallization screen. This can be the starting point for crystallization for these proteins so that their structure can be determined.
If the crystal produced was not well-formed or large enough, scientists can still use the information to help them better determine the conditions necessary to create a well-formed crystal. For example, they may learn that Protein X and Condition A resulted in a micro crystal, and Protein A and Condition Z resulted in a micro crystal as well. Based on this information, they can then run additional experiments to deduce what conditions need to be optimized to create a larger and well-formed crystal.
Analyzing the results from this experiment will also lead to better understanding the underlying principles of protein crystallography. For the first time, a comprehensive crystallography image analysis will be done, which was impossible before due to computational complexity. In turn, CrystalVision will be improved to provide faster and more accurate image classification.
Improving the protein crystallography pipeline will enable researchers to determine the structure of many cancer-related proteins faster. This will lead to improving our understanding of the function of these proteins, and enable potential pharmaceutical interventions to treat this deadly disease.
October 2015 Update
We continue to analyze the millions of protein-crystallization images that you processed as part of Help Conquer Cancer project, with the end goal of gaining insight into the crystallization process. In turn, this will enable to crystalize cancer- (and other disease-) related proteins, determine their structure, function, and design drugs as needed.
Read More
We continue to analyze the millions of protein-crystallization images that you processed as part of Help Conquer Cancer project, with the end goal of gaining insight into the crystallization process. In turn, this will enable to crystalize cancer- (and other disease-) related proteins, determine their structure, function, and design drugs as needed.
Read More

June 2015 Update
We continue to analyze the millions of protein-crystallization images processed by World Community Grid volunteers
Read More
We continue to analyze the millions of protein-crystallization images processed by World Community Grid volunteers
Read More
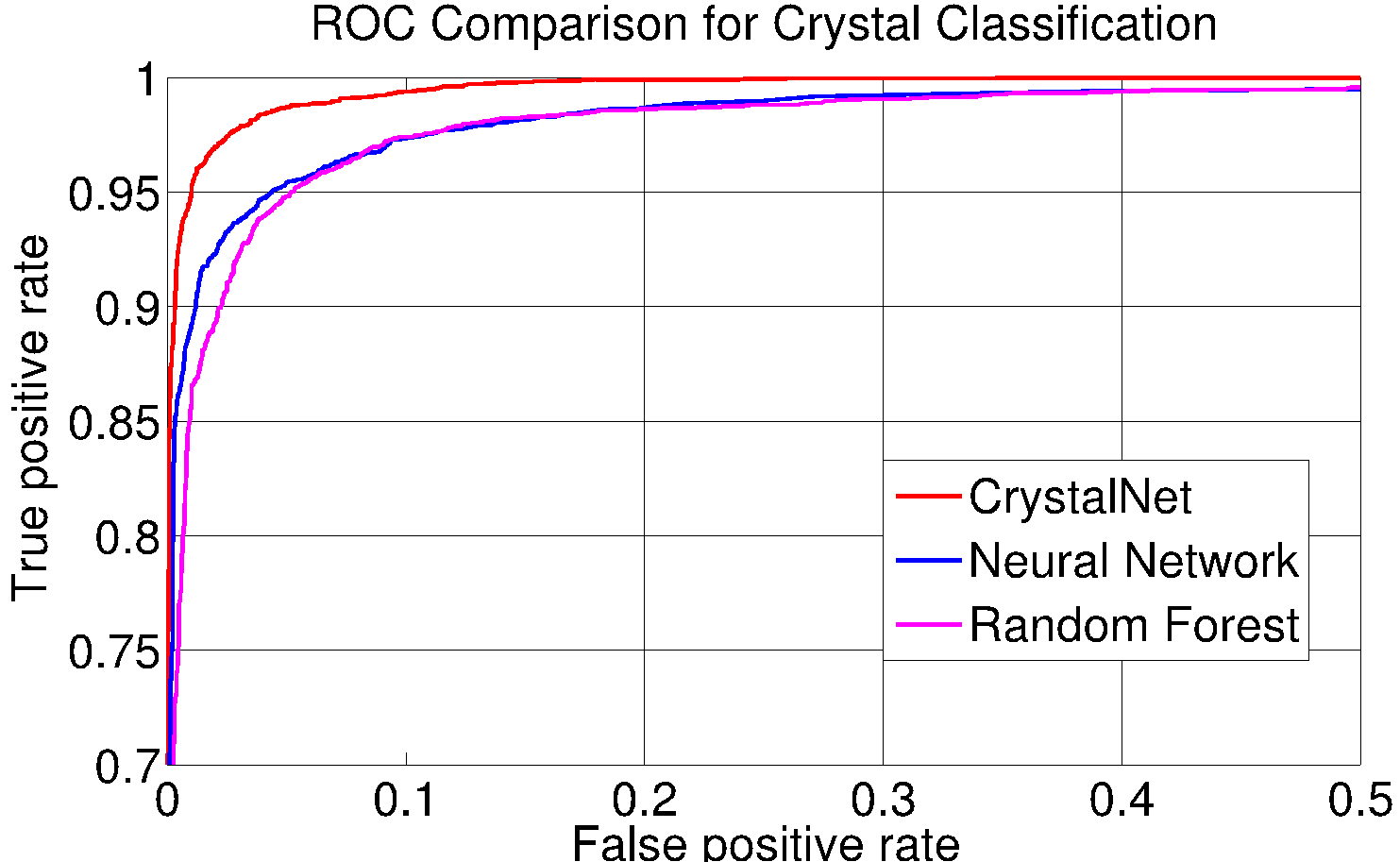
December 2014 Update
Introducing deep convolutional neural networks - CrystalNet - for an accurate and efficient classifier for protein crystallization images
Read More
Introducing deep convolutional neural networks - CrystalNet - for an accurate and efficient classifier for protein crystallization images
Read More
April 2014 Update
Analysis of HCC results is in progress, and there are some exciting results we will be reporting on next time. However, over the last year considerable energy and resources were devoted to the new project on WCG –Mapping Cancer Markers project (MCM), and other cancer-gene-signature projects, in which our research group is involved.
Read More
Analysis of HCC results is in progress, and there are some exciting results we will be reporting on next time. However, over the last year considerable energy and resources were devoted to the new project on WCG –Mapping Cancer Markers project (MCM), and other cancer-gene-signature projects, in which our research group is involved.
Read More
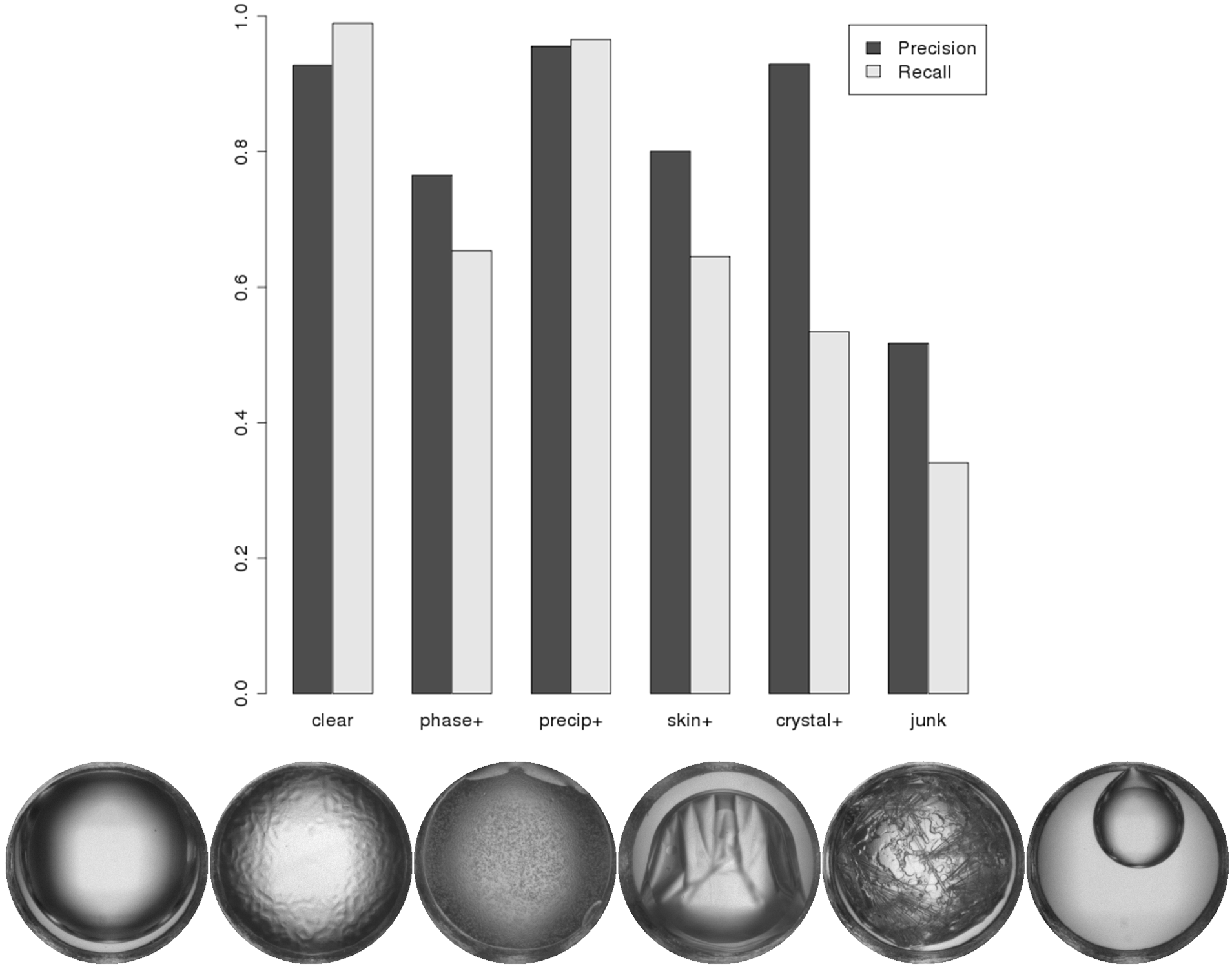
April 2013 Update
Introducing a new set of binary classifiers to complement our existing suite of Random Forest classifiers -- substantially improving precision and recall of crystals and junk.
Read More
Introducing a new set of binary classifiers to complement our existing suite of Random Forest classifiers -- substantially improving precision and recall of crystals and junk.
Read More
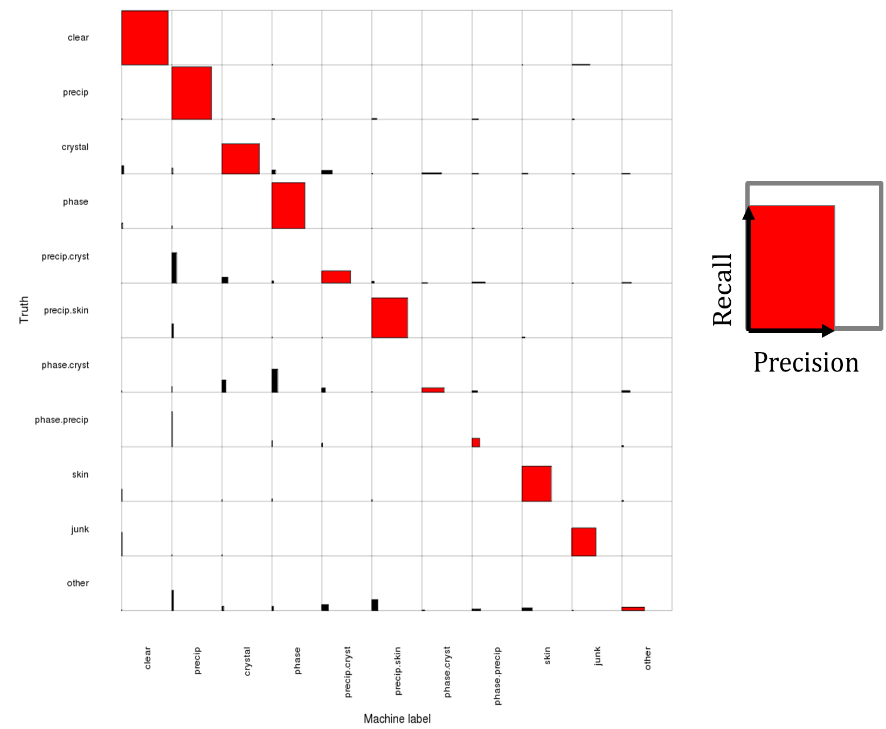
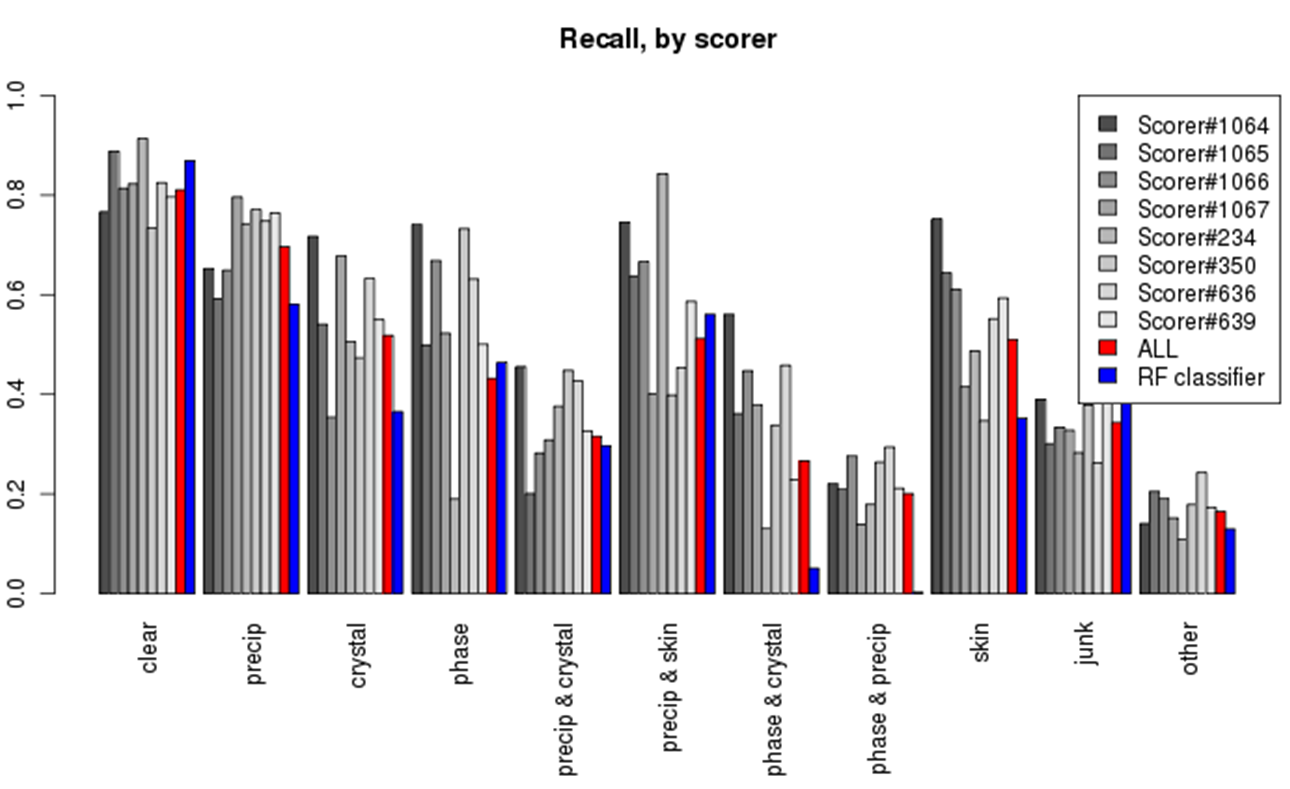
November 2012 Update
Introducing an improved 11-way logistic regression classifier: Recall is improved on the most common classes, clear (98.8%) and precipitate (96.0). Precision is improved on crystal (74.9) and other less-common classes
Read More
Introducing an improved 11-way logistic regression classifier: Recall is improved on the most common classes, clear (98.8%) and precipitate (96.0). Precision is improved on crystal (74.9) and other less-common classes
Read More
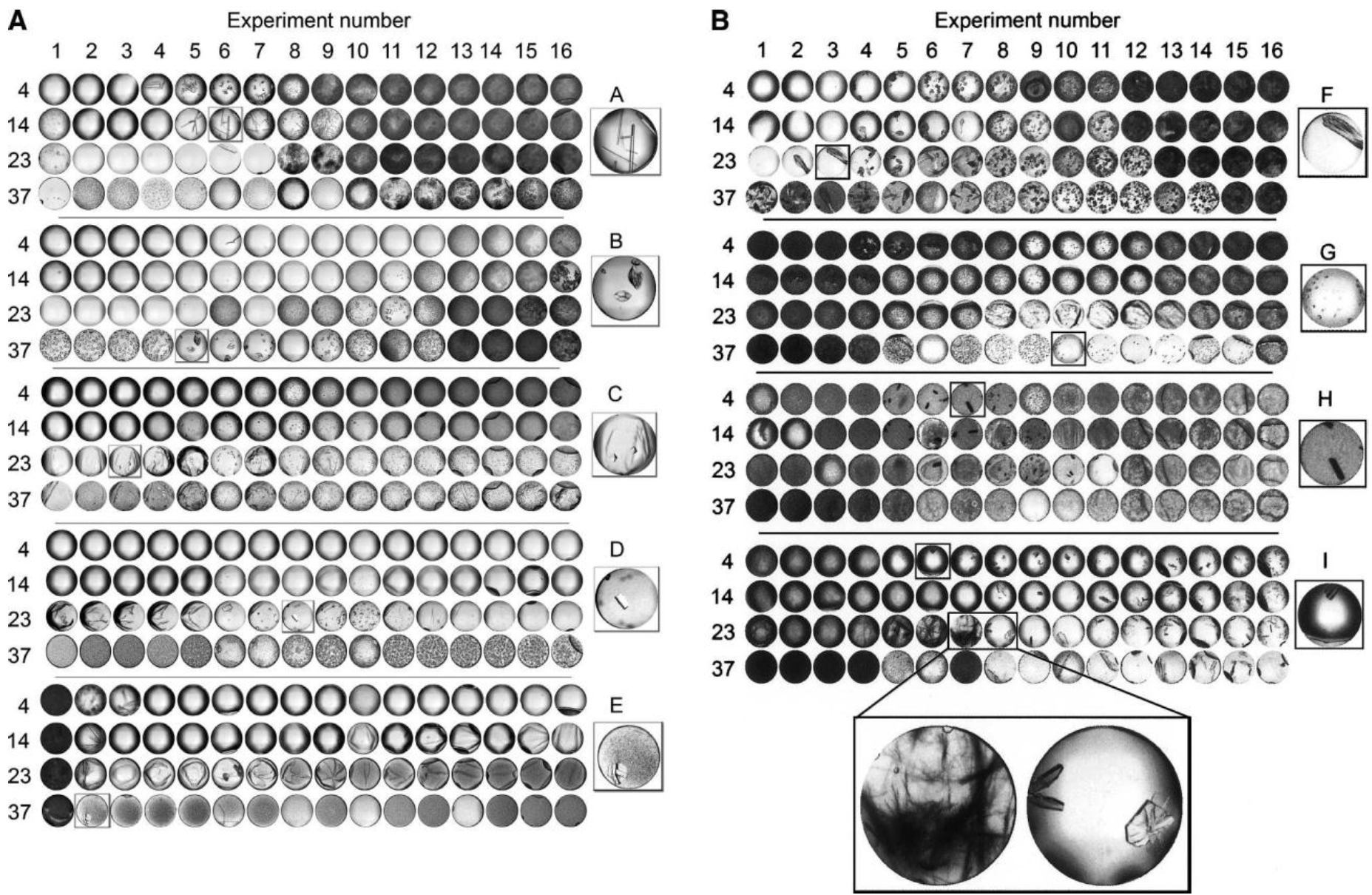
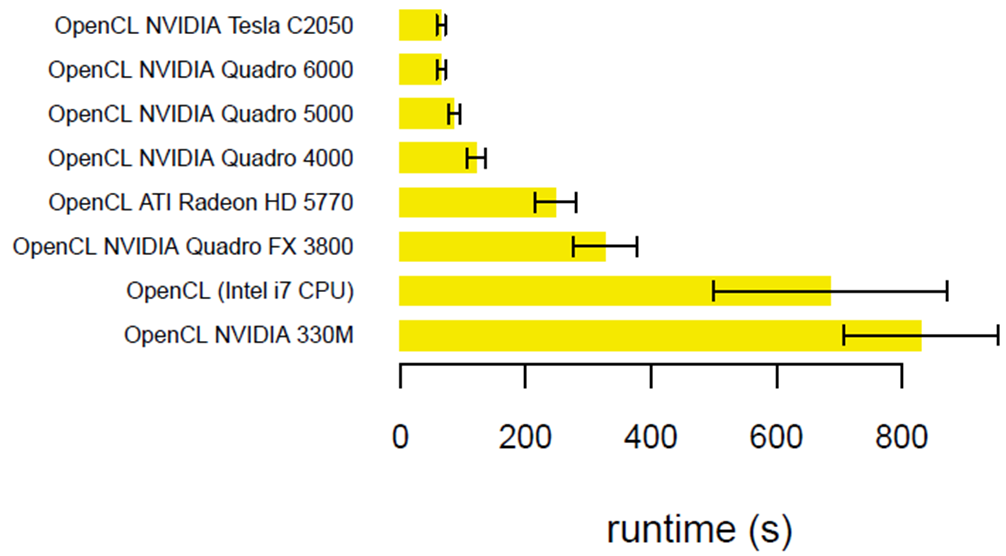
September 2011 Update
Introducing a GPU-accelerated HCC code - CPU runtime averages 4092 seconds (single threaded) on an Intel Xeon, but only 65 seconds on an NVIDIA Tesla C2050.
Read More
Introducing a GPU-accelerated HCC code - CPU runtime averages 4092 seconds (single threaded) on an Intel Xeon, but only 65 seconds on an NVIDIA Tesla C2050.
Read More
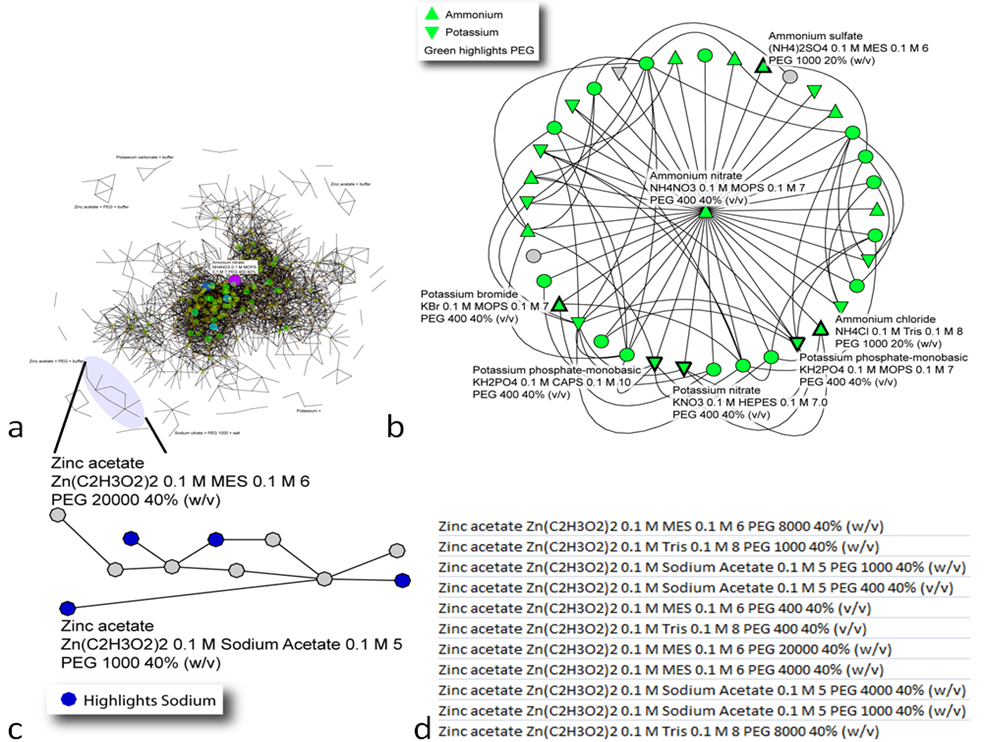
October 2010 Update
Network-based analysis of relationship among crystallization cocktails and proteins.
Read More
Network-based analysis of relationship among crystallization cocktails and proteins.
Read More

March 2010 Update
The system successfully recognizes 80% of crystal-bearing images and 95% of clear drops.
Read More
The system successfully recognizes 80% of crystal-bearing images and 95% of clear drops.
Read More
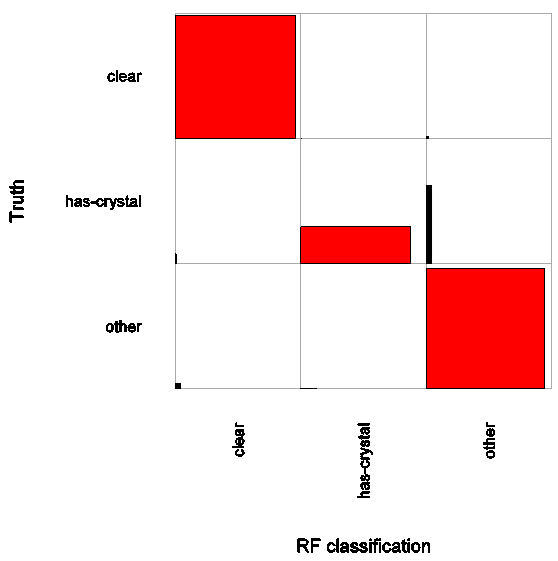
October 2009 Update
Impressive precision and recall especially for clear and "other" category makes the computer system equal or better to the human expert.
Read More
Impressive precision and recall especially for clear and "other" category makes the computer system equal or better to the human expert.
Read More

April 2009 Update
25% of our work units have been processed on the WCG – representing 3 million crystallization trials on over 2,000 proteins.
Read More
25% of our work units have been processed on the WCG – representing 3 million crystallization trials on over 2,000 proteins.
Read More
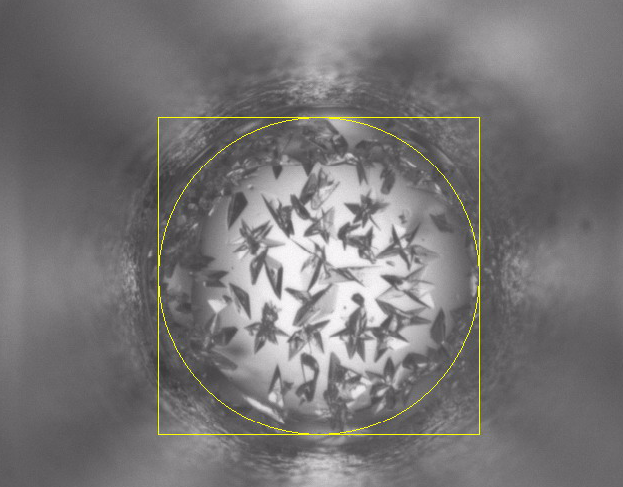
October 2008 Update
The results show improved detection of crystals as a whole (69% sensitivity, 78% specificity when combining crystal, crystal/phase, crystal/precip classes).
Read More
The results show improved detection of crystals as a whole (69% sensitivity, 78% specificity when combining crystal, crystal/phase, crystal/precip classes).
Read More

May 2008 Update
During the first phase of the project, we processed a well-characterized set of 165,416 images. We are in the process of optimizing features across wide range of individual image categories.
Read More
During the first phase of the project, we processed a well-characterized set of 165,416 images. We are in the process of optimizing features across wide range of individual image categories.
Read More

January 2008 Update
Although we have over 84,000,000 images to process, we have focused on a well-characterized set of 85,261 images first, to improve the CrystalVision software.
Read More
Although we have over 84,000,000 images to process, we have focused on a well-characterized set of 85,261 images first, to improve the CrystalVision software.
Read More

2007 Welcome Intro
For the first time, a comprehensive crystallography image analysis will be done, which was impossible before due to computational complexity
Read More
For the first time, a comprehensive crystallography image analysis will be done, which was impossible before due to computational complexity
Read More
All contents copyright Jurisica Lab, Krembil Research Institute, UHN. Last modified March 2024





